
We hear of groundbreaking scientific advances in medicine every day. But still there are some questions that even the latest technology falls short of answers. They vary from finding a vaccine for a minuscule virus to finding a cure for a complex disease.
Similarly, for decades scientists have been trying to find a cure for Huntington Disease, a rare but serious disease affecting thousands across the globe.
How far are we into the journey of finding a treatment for HD? Is there light at the end of the tunnel after all? Is there an end to this long agonizing waiting game in sight? Here’s an attempt to find out answers to these burning questions. Let’s dive in.
Table of Contents
What is Huntington Disease?
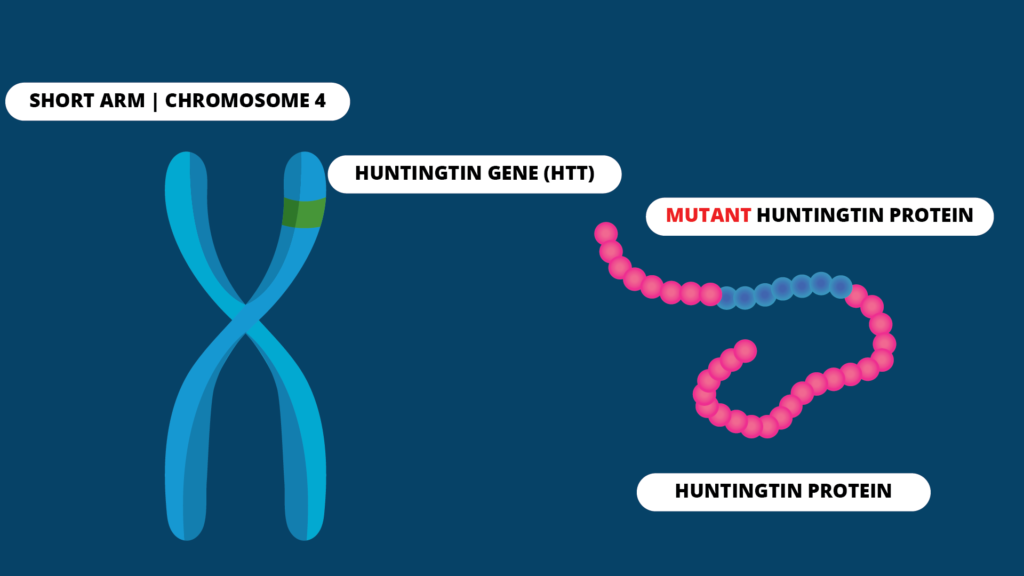
Huntington Disease (HD), is a rare genetic disorder which affects the brain and its functions in a progressive manner. It is caused by a genetic mutation occurring in the short arm of chromosome 4 which acts as the blueprint for the Huntington protein production. This protein is also called the Huntingtin protein.
All humans have two copies of this gene. It has a section called trinucleotide repeat, which is a sequence of three DNA bases, Cytosine (C), Adenine (A), and Guanine (G), repeated multiple times. In patients with HD, the sequence gets repeated several times so that a protein longer than the usual is produced. Depending on the number of times, this CAG sequence gets repeated, the age of onset of symptoms of HD can vary. It typically presents between the ages of 30-50 years.
The mutation of the gene is dominant, which means there’s a 50:50 chance of you getting the disease if either one of your parents has also got it. Moreover, it is a disease with complete penetrance which means that all disease carriers will develop the disease at some point or another during their lifetime.
Symptoms
Unlike the normal Huntington protein, the protein produced by the mutated gene is toxic to the brain cells. Therefore, it can lead to neuronal death, causing damage to signal pathways in the brain. A variety of neurological symptoms occur as a result. They are mainly motor, cognitive, or psychiatric in nature.

Chorea is considered as one of the most striking motor symptoms of HD. It is characterized by excessive involuntary movements that last a short duration and appear to be semi-purposeful. Besides, people affected by HD often manifest cognitive symptoms like impairment in memory, communication, and attention. Psychiatric symptoms like depression, aggression and dementia are also not so uncommon.
What are the treatments available?
Despite spending years conducting a great number of researches, scientists still have not been able to name a definite cure for HD yet. However, several treatment modalities have come into light over the years, mainly to improve the symptoms of the disease and to delay disease progression.
Symptomatic Therapies for Motor Symptoms
Several medications are used to overcome movement problems like chorea, especially when it starts to hinder day to day lifestyle with increased risk of falling or getting injured. These drugs mainly target the activity of neurotransmitters like dopamine, NMDA and glutamate, which are small chemical compounds that act as messengers between neurons. When there is an over-activity or increased amounts of these neurotransmitters, they tend to over-excite areas of brain responsible for movement control. As a result, abnormal chorea like movements are generated. Tetrabenazine is one such drug, commonly used to reduce chorea like movements by reducing dopamine activity in the brain. Other medications include, Amantadine and Riluzole which have anti- NMDA and anti-glutamate activities respectively.
Symptomatic therapies for behavior symptoms
Symptoms like depression are overcome with the help of medications like Selective Serotonin Reuptake Inhibitors (SSRIs) such as citalopram and Stimulating SSRIs such as fluoxetine.
To tackle symptoms like aggression, irritability, and psychosis, typical and atypical antipsychotics like risperidone, olanzapine, or haloperidol are utilized. In addition, mood stabilizers like benzodiazepines and lithium are used to treat mood changes depending on the clinical situation.
Disease-Modifying Therapies
Ever since the groundbreaking discovery of the HD causing gene in 1993, scientists have been thriving hard to find a cure for HD at the genetic level. However, the efforts in developing a drug to alter the progression of the disease has mostly been nothing short of unsatisfactory. After several years of waiting with many ups and downs, there seems to be new hope now as several clinical trials targeting the gene have started to show successful preliminary results.
Huntingtin-Lowering Therapies
As you must be well aware by now, the toxic protein called, Huntingtin which is produced by mutated HD gene, is the culprit wreaking havoc inside the brain causing the disease in the first place. Several studies have discovered that this mutated protein is responsible for dysregulation of brain functions. So, it is simple logic that finding a way of tackling down this trouble making toxic protein will be a key to finding a cure for HD. Hence comes the concept of huntingtin lowering therapy into play.
DNA indeed acts as the template for creating proteins in cells. However, DNA does not directly produce proteins. There is a series of events and other intermediate participants that contribute to it.
It is a type of molecules called RNA which copies information (transcription) on DNA and transport them to the protein factory in the cell for protein production. Since, these RNA molecules carry messages and instructions from DNA molecules, they are simply known as messenger RNAs or mRNAs. In a therapeutic point of view, if these messenger molecules are destroyed, before they get a chance to deliver the message about mutant protein production, it could be a potential way of reducing disease progression.
In summary, the types of huntingtin lowering therapies are threefold. They either target the mutated gene at the DNA level, destroy messenger RNAs at transcription level or beyond, or target and destroy the toxic protein at the cellular level. They are collectively known as Gene Silencing as well.
DNA Targeting Approaches
The DNA targeting approaches modify and edit the genome to avoid the production of mutant proteins. This method requires special tools called DNA binding elements and effectors. As the names would imply, DNA-binding elements bind to the specific mutated places in the DNA so that the effectors can cut and repair the affected gene.
Zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and Cas9 or other RNA-guided bacterial nucleases are the three main types of DNA-binding agents. They differ from each other in their mechanism of DNA binding and mode of action.
Genome editing technology is still in its infancy stage. If done correctly, it will be a one-time treatment with long-term positive effects. However, there are some major setbacks. It targets only a limited area of the brain. Then there’s the risk of other areas with CAG-sequence repeats being affected as well. Since the procedure is invasive and irreversible a great deal of caution has to be applied.
Even though this method is pursued as a potential cure for HD with promising results from trials done up to now, extensive pre-clinical testing is mandatory before starting human trials.
HD-GeneTRX-1 by UniQure
UniQure is a pioneer in gene therapy and it conducts a popular study named HD-GeneTRX-1 at the moment. The US Food and Drug Administration (FDA) has already promoted their product, AMT-130, for the status of an “Investigational New Drug”, which is a huge achievement as far as drug trials are concerned.
The way AMT-130 drug works is advanced yet quite interesting. They make use of a small, harmless virus called adeno-associated virus or AAV. They use gene technology to remove DNA in the virus only to replace them with DNA having information and instructions that are rather useful.
Viruses have this amazing ability to sneak into host cells and reprogram their DNA with their own new DNA. So, the scientists are making use of AAV to carry information necessary to make a type of molecules called micro-RNA. These micro-RNA molecules have the ability to bind to messenger RNA of HTT protein and destroy them before they convey the message necessary for toxic protein production. Pretty cool, isn’t it?
The AMT-130 drug has all the elements necessary to achieve this. It should be delivered via brain surgery, and requires extreme degree of precision to deliver the right amount of viral load to the right parts of the brain. So, this is not exactly an easy job to do.
Up to now the studies done on animals have shown promising results and now they have moved onto conducting human trials. However, it is too soon to tell when this method will exactly make its appearance in routine clinical practice for now.
RNA Targeting Approaches
These approaches mainly target messenger RNA molecules by either disrupting the information copying phase or destroying them altogether. This leads to reduction in the mutant protein production at the end.
Small molecules
Small molecules approach includes using a drug which can be taken orally as a pill to destroy mRNA responsible for producing mutant HTT. It is an innovative approach and sounds a lot more appealing than cutting into brain for drug administration. Several companies are working on the subject at the moment.
PTC518, a small molecule developed by PTC Therapeutics, has been successful in lowering the Huntingtin protein throughout the brain in animal models in a uniform way. PTC518 is considered as the only small molecule which can target Huntington gene expression with great precision. Now they have already announced human trials for the drug.
Novartis is another company following a similar approach with a small molecule, but with an already existing drug called Branaplam.
Branaplam was initially used to treat another childhood genetic disease called Spinal Muscular Atrophy (SMA). They have observed that when children with SMA are treated with this drug, their huntingtin levels are also reduced.
Based on these findings, Novartis began testing Branaplam for HD on animal and HD-cell models and both have manifested successful results. The drug has even passed a safety trial conducted using a small number of healthy adults. Now the company is planning to conduct human trials at a larger scale in 2021. If these trials succeed, it means that we’ll be having a familiar oral drug in town to combat HD, hopefully in the very near future.
Antisense oligonucleotides or ASOs
Of all the huntingtin-lowering technologies that exist, ASOs are the ones making headlines quite often. In fact, their discovery is considered nothing short of groundbreaking.
Antisense oligonucleotides or ASOs are small fragments of DNA having a chemically modified structure. They are capable of freely entering into cells. Their action is to destroy messenger RNA having information on making toxic HTT proteins.
In the year 2015, The IONIS Pharmaceuticals together with another giant in the industry, Roche, developed an ASO drug for HD, called “IONIS-HTT Rx”. Then they developed a study to determine whether the drug is safe for administration or not.
This particular drug has to be delivered via a procedure called lumbar puncture, by piercing the base of spine, to make sure the drug reaches the brain. Although the procedure sounds alarming, it is neither novel nor dangerous but on contrary quite routine.
This study included a placebo as well, which means some participants were not expose to the drug. A multiple ascending dose regimen was used to increase the safety of the study. This means the early participants were given a low dose while those who joined later were given a higher dose allowing time for monitoring side effects.
The exciting news reached in 2017, when IONIS revealed in a press release that the study has shown dose-dependent reduction in mutant huntingtin protein. This is considered as the next greatest achievement in HD research after the discovery of the HD gene in 1993. Most importantly, the drug has not manifested any safety concerns in any of the participants. However, it is not yet clear whether this amount of reduction in HTT protein is enough to reverse or reduce HD symptoms.
IONIS and Roche are now in the process of expanding their research to determine whether IONIS-HTTRx, now known as Tominersen, has the potential to reduce disease progression or not. For this purpose, they have currently launched the study named, GENERATION-HD1, with participation of nearly 1000 participants at more than 100 sites across 19 countries in the world. Its results are expected to be available by 2022.
Scientists have very high expectations for Tominerson as a definitive treatment for people having HD. If successful, Tominerson might as well as be the wonder drug we all have been waiting for.
The waiting game is almost over
After decades of waiting with frustration and agony, finally there’s hope for people with HD now. Multiple companies explore multiple ways to tackle the mutant HTT protein. Besides, multiple interventions and strategies are developed every day with the common objective of defeating HD. For the first time in history, it will not be decades, but years before a treatment will be among us. The light at the end of the tunnel is far within our reach now and we are closer to winning the battle against HD than we have ever been before.
References
- Ghosh R, Tabrizi SJ. Clinical aspects of Huntington’s disease. In Behavioral Neurobiology of Huntington’s Disease and Parkinson’s Disease 2013 (pp. 3-31). Springer, Berlin, Heidelberg.
- Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron. 2019 Mar 6;101(5):801-19.
- Colpo GD, Rocha NP, Stimming EF, Teixeira AL. Immunomodulatory Strategies for Huntington’s Disease Treatment. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders). 2017 Oct 1;16(8):936-44.
- Huntington’s Disease Association Website https://www.hda.org.uk/
- HD BUZZ-Huntington’s disease research news https://en.hdbuzz.net/