
Table of Contents
CASE SCENARIO
Matt, an 8-year-old male, presented with persistent jaundice, intermittent itching, and the passage of pale-coloured stool. Onwards 10 days after birth, all these signs were noted. Matt’s parents noticed a slowly growing lump over the right upper quadrant of his abdomen for the past 6 years.
Additionally, he had a 4-year history of an intermittent productive cough accompanied by sneezing, rhinorrhea, and occasional fever. Episodes of productive cough mainly occurred during the winter and rainy seasons and generally lasted 4–5 days and improved following treatment with expectorant and anti-allergic medication. He had intermittent nose bleeding, post-traumatic violaceous-red patches that occasionally appeared at the site of the trauma, and recurrent post-traumatic fractures of his upper limbs after mild trauma for the previous 4 years. He has also had difficulty with night vision for the past 3 years.
No prior history of altered sensorium, blood vomiting, or passage of black stools existed for our patient. He was born 2 weeks after the expected delivery date, with a low birth weight of 2kg, delayed growth and development, and gained weight poorly throughout infancy. The patient had no prior history of asthma or tuberculosis.
Based on his family history, there were no similar illnesses, asthma, tuberculosis, liver conditions or consanguineous marriages. He has two older, asymptomatic siblings who are healthy.
On physical examination, his height was 110 cm, weight 12 kg, and a BMI of 9.9 kg/m2. Examination of his skin revealed he had icterus, pallor, clubbing, and scratch marks over his skin. There was no testicular atrophy, gynecomastia, purpura, palmer erythema, spider nevi, or pedal edema.
On head and neck examination, the patient had distinctive facial features in the form of a triangular face with a broad forehead, deeply set eyes, prominent ears, a small pointed chin, and an elongated nose with a bulbous tip.
Abdominal examination revealed enlargement of the left lobe of his liver. The liver was firm in consistency with an irregular margin and slightly nodular surface. His liver span was 9 cm along the right mid-clavicular line, and his spleen was palpable 4 cm below the left costal margin and firm in consistency.
Ophthalmologic examination demonstrated Axenfeld anomaly, posterior embryotoxon, eccentric pupils, and increased intraocular pressure.
Respiratory and neurological examinations were normal.
On his laboratory examinations, he tested negative for anti-hepatitis C antibodies and hepatitis-B surface antigen.
On imaging studies, his wrist X-ray revealed severe osteopenia, and his chest X-ray revealed cardiomegaly. Tricuspid regurgitation and pulmonary hypertension were detected by two-dimensional echocardiography. His abdomen was examined using ultrasonography, which revealed a mildly enlarged left lobe, a shrunken right lobe with coarse hepatic parenchyma, and poorly visible hepatic and portal veins. Liver biopsy revealed portal tracts with a conspicuous absence of interlobular bile ducts adjacent to the hepatocytes, a portal area with mild inflammation, some degree of fibrosis, and scattered nodules.
The physician suspected Alagille syndrome, along with bile duct paucity and confirmed the diagnosis through molecular genetic testing.
WHAT IS ALAGILLE SYNDROME?
Alagille syndrome (ALGS), also known as Alagille-Watson syndrome or arteriohepatic dysplasia, is a rare autosomal dominant condition with multiorgan involvement. This condition was first described by the French pediatrician Daniel Alagille in 1969, hence the name “Alagille syndrome”.
WHAT CAUSES ALAGILLE SYNDROME AND IN WHOM?
Alagille syndrome affects both male and female populations equally. Currently, two genes are identified in which mutations of their expression can result in Alagille syndrome; these are the JAG1 and NOTCH2 genes.
The majority of the cases are linked with JAG1 (more than 95%), and the resulting mutations are either complete gene deletions, frameshift mutations, nonsense, or missense mutations.
These genes are responsible for giving instructions for encoding proteins that participate in the Notch signaling pathway. Notch signaling pathway is vital in building body structures during embryonic development. Mutations in these genes will lead to non-functional proteins in the notch signaling cascade, ultimately leading to poor and incomplete embryonic development.
This, in turn, causes the characteristic signs seen with this condition, and will be discussed in detail later in our article.
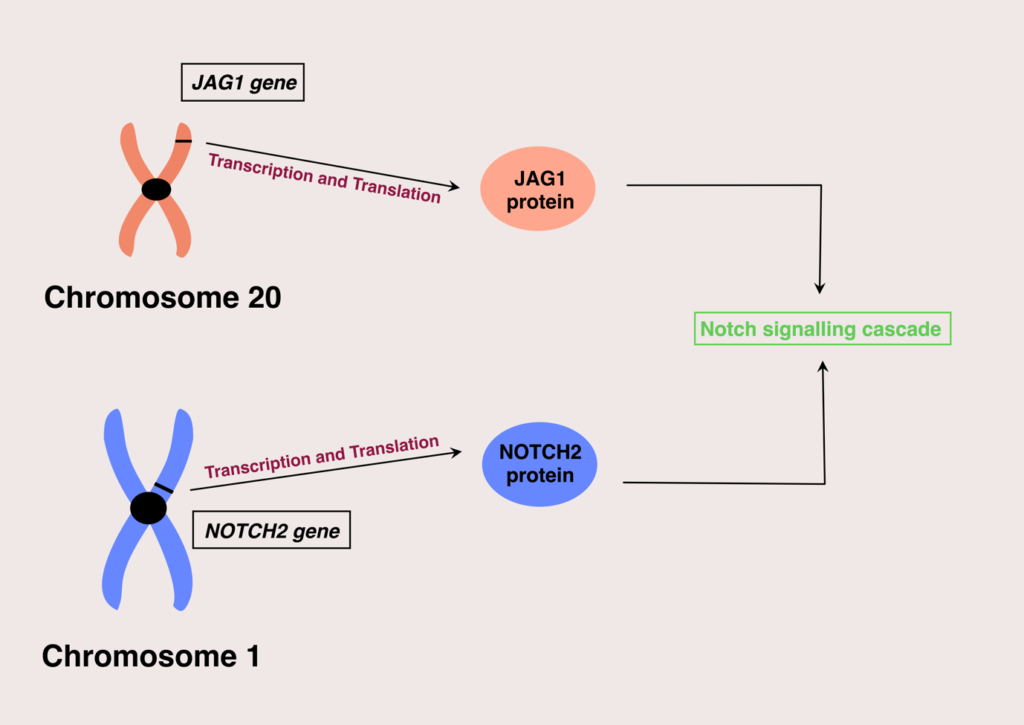
As mentioned in the beginning, Alagille syndrome is passed along in an autosomal dominant pattern, with variable expression among individuals. This means a child has a fifty per cent chance of inheriting the disease if any one of its parents is affected.
The majority of the children diagnosed with Alagille syndrome had new mutations in their JAG1 OR NOTCH2 genes without having any family history, and the others inherited the syndrome from one of their parents. So, this means not having a family history doesn’t necessarily mean that an individual is safe from this condition.
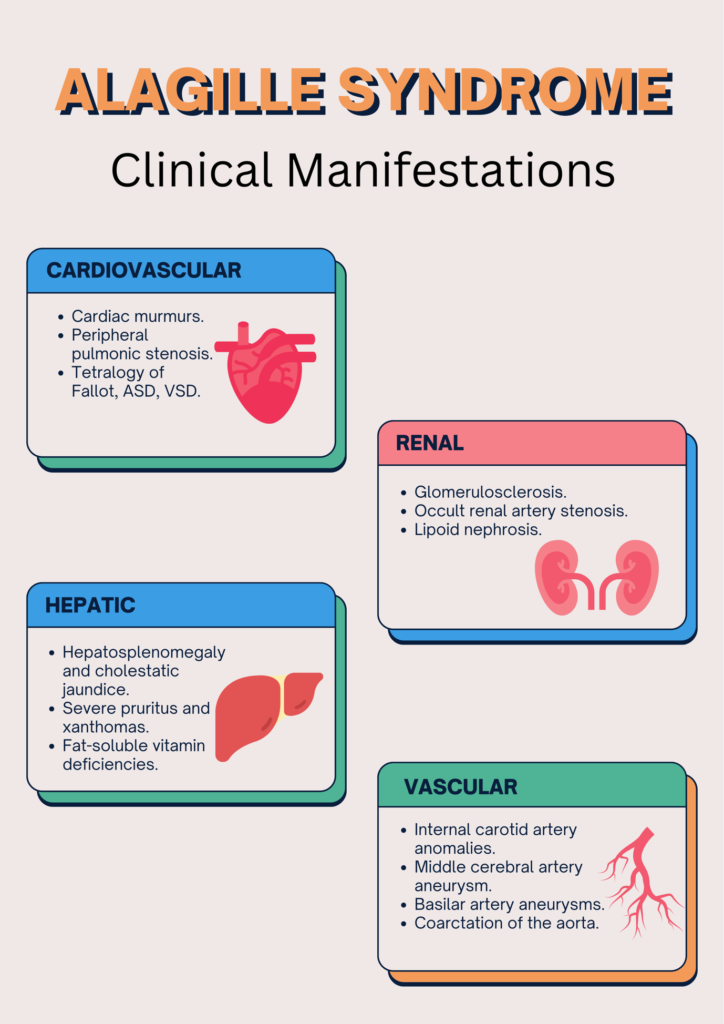
WHAT ARE THE CLINICAL MANIFESTATIONS?
Owing to the multisystem involvement, the presentation of Alagille syndrome varies from individual to individual.

WHAT ARE THE INVESTIGATIONS NECESSARY TO EVALUATE ALGS?
By now, you might have figured out that hepatic disease is a crucial feature of Alagille syndrome. Therefore, evaluating suspected patients for chronic liver disease is a significant part of the investigative studies.
Liver biopsy can reveal interlobular bile duct paucity and suggest chronic cholestasis in ALGS patients, making it a valuable tool in evaluating ALGS patients.
Even though the hepatic disease is a key feature, this finding does not always occur in infants suffering from Alagille syndrome. Therefore, additional laboratory and imaging studies should be conducted to aid in the diagnosis of ALGS. These include testing the parameters of function and differential diagnoses.
Frequently, liver function deterioration and fat-soluble vitamin deficiencies are encountered with Alagille syndrome. These can be detected using a plethora of blood tests.
The levels of gamma-glutamyl transpeptidase (GGT) and alkaline phosphatase are generally elevated, and with increased amounts of cholic and chenodeoxycholic acids, serum bile acids are also significantly elevated.
Prothrombin time (PT) or activated partial thromboplastin time (aPTT) prolongation is commonly seen, along with hypercholesterolemia and hypertriglyceridemia. This usually points out to chronic cholestasis in ALGS patients.
Excluding differential diagnoses is also an essential part of diagnosis, which will be covered in detail in the subsequent sections.
Imaging studies, in addition to laboratory tests, are used to evaluate associated malformations and rule out differential diagnoses. An array of studies are used for this purpose.
Abdominal ultrasonography is used to examine the hepatobiliary tree and the hepatic parenchyma and to screen for renal anomalies.
We can employ tests including endoscopic retrograde cholangiopancreatography (ERCP), intraoperative cholangiography, dimethyl iminodiacetic acid (HIDA) scanning, and magnetic resonance cholangiopancreatography for further clarifications of the biliary anatomy.
Skeletal abnormalities such as butterfly vertebrae are screened via X-ray.
In order to screen for cardiovascular abnormalities, ECG should be used.
An ophthalmologic examination should screen for posterior embryotoxon, Axenfeld anomaly, and retinal changes.
WHAT ARE THE DIFFERENTIAL DIAGNOSES?
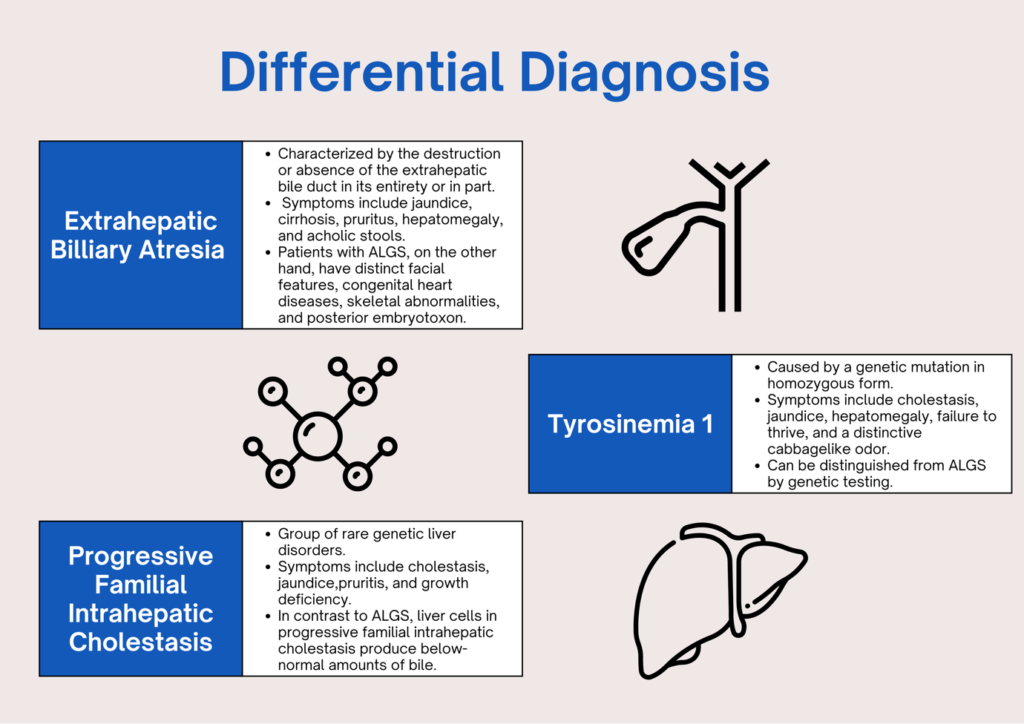
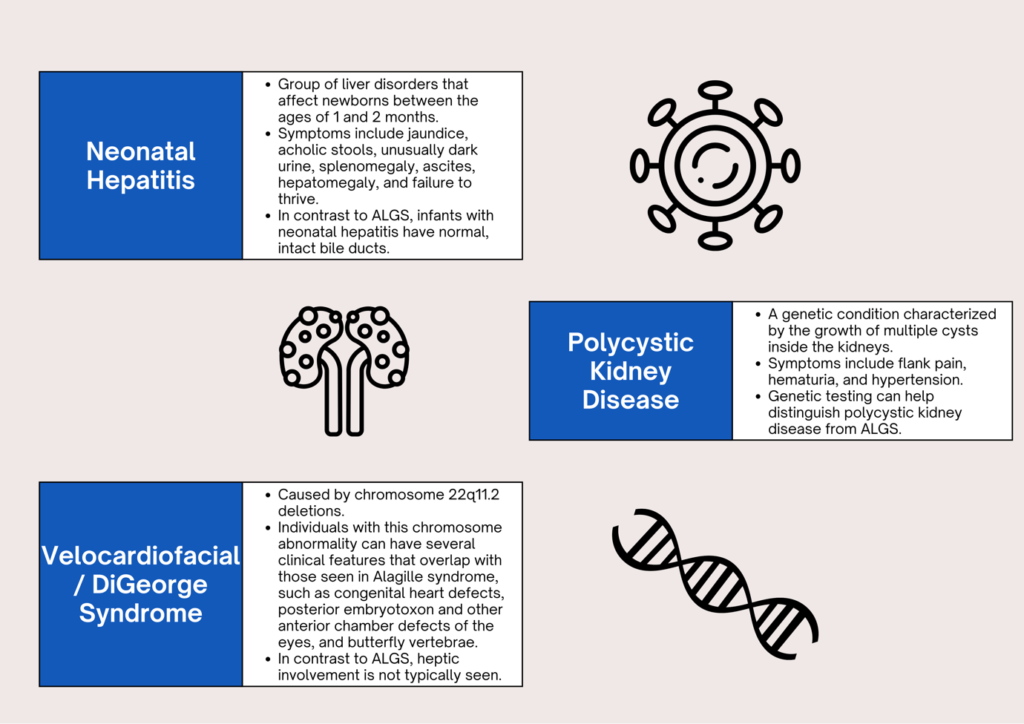
HOW DO WE DIAGNOSE ALGS?
Diagnosis of Alagille syndrome can be very tricky, owing to the variable nature of its presentation. Some patients are identified at birth with the classical signs of Alagille syndrome, some may present with the symptoms at a later age, while some may not show any symptom or sign at all, but still, a careful examination may diagnose ALGS following the identification of a case in their family. Therefore, the diagnosis should be based on the presence of characteristic symptoms, a detailed patient history, a thorough clinical evaluation, and a battery of investigative tests.
Five hallmark clinical findings are identified in Alagille syndrome, and these include cholestasis with bile duct paucity, cardiac abnormalities, skeletal anomaly (mainly butterfly vertebrae), ophthalmologic abnormality (posterior embryotoxon), and distinctive facial features. So, if a patient presents with three or more of these clinical findings, the physician should confirm the diagnosis and take the necessary steps.
The physician can arrive at a definitive diagnosis by molecular genetic testing, which tests for genetic mutations in JAG1 or NOTCH2 genes. But this may not be true for everyone with the syndrome because genetic testing may not reveal mutations in these individuals. So, the physician should be skilled enough to make educated decisions based only on the patient history and investigation results in these situations.
WHAT ARE THE TREATMENT OPTIONS AVAILABLE?
Unfortunately, even with our extensive knowledge of the disease’s characteristics, there is no known cure for ALGS. Patients with Alagille syndrome should receive multidisciplinary treatment through coordinated efforts from a team of clinical specialists due to the variable nature of its presentation. Treatment is focused on the specific signs and symptoms that individual patient experiences and can be broadly categorized as medical and surgical.
MEDICAL MANAGEMENT
Medical management is aimed at improving biliary flow, reducing blood cholesterol levels, maintaining normal growth and development, preventing complications, and improving the overall quality of life.
Bile is an essential component in the absorption of fat-soluble vitamins. Therefore, cholestasis may lead to fat-soluble vitamin (vitamin K, A, D, and E) deficiencies in Alagille syndrome. So, supplementation of these vitamins in appropriate doses is indicated. Sometimes, zinc deficiency is observed and can be easily replaced via oral supplementation.
In addition to vitamin deficiencies, caloric insufficiency is also seen among patients with Alagille syndrome. Thus, some affected children may need to ingest additional calories through a nasogastric or a gastrotomy tube.
Improving the bile blow, thereby managing cholestasis, is essential in any patient with Alagille syndrome. Cholestasis leads to some problematic conditions, such as pruritus and xanthomas, which can affect one’s quality of life.
Ursodiol (Ursodeoxycholic acid) is commonly administered to improve the bile flow and relieve the symptoms of pruritus and xanthoma formation.
Apart from general treatment for cholestasis, pruritus is also treated with antihistamines (hydroxyzine and diphenhydramine), cholestyramine, and rifampin. Newly approved drugs such as ileal bile acid transport (IBAT) inhibitors (maralixibat) are also used to treat cholestatic pruritus in Alagille syndrome.
Xanthomas are cholesterol deposits in the skin and are also treated with cholestyramine, which reduces blood cholesterol levels.
SURGICAL MANAGEMENT
Surgical treatments are reserved for patients who are resistant or do not respond to drug therapy and those with life-threatening complications.
Partial internal biliary diversion (PIBD) and partial external biliary diversion (PEBD) are surgical procedures used to reduce the reabsorption of bile acids from the terminal ileum, thus reducing symptoms of pruritus and xanthoma formation.
Orthotopic liver transplantation should be considered in more sinister cases where the disease may progress to liver failure.
Even though the bulk of the treatment is towards managing liver diseases, we should not forget the additional complications involving numerous other organ systems.
The patient should consult a cardiologist if severe cardiovascular abnormalities such as patent ductus arteriosus (PDA), tetralogy of Fallot, etc. or vascular anomalies including aneurysms and stenoses are encountered.
Posterior embryotoxon, Axenfeld anomaly, and other eye diseases should be managed accordingly by an ophthalmologist and if renal insufficiency is observed, it should be brought to a nephrologist’s attention right away.
Additionally, it is advised that affected individuals and their families seek genetic counselling.
REVISITING THE PATIENT
Now, let’s turn our spotlight back onto the patient.
Our patient was treated with Ursodiol, vitamin A and D3 tablets, calcium, and a multivitamin with zinc. Although Matt’s parents were instructed to attend routine follow-up appointments, they never showed up. Unfortunately, 1 year after receiving the final diagnosis, Matt passed away from severe hematemesis.
IN CONCLUSION
Alagille syndrome is a rare genetic disorder that affects the liver, heart, bones, and other body organs. It is mainly characterized by bile duct paucity, among other organ system abnormalities. Some patients may experience mild symptoms, while others have severe symptoms that affect every aspect of their lives.
It is diagnosed by a physician through a multitude of tests and can be confirmed with genetic testing. The only treatment for Alagille syndrome is to treat symptoms as they occur, and currently, there is no known cure for the disease.
Alagille syndrome has an undetermined long-term prognosis, whilst congenital heart disease, hepatic cirrhosis, and renal abnormalities are the most common causes of death.
RESOURCES
- Tretter, J.T., McElhinney, D.B. (2018). Cardiac, Aortic, and Pulmonary Vascular Involvement in Alagille Syndrome. In: Kamath, B., Loomes, K. (eds) Alagille Syndrome. Springer, Cham. https://doi.org/10.1007/978-3-319-94571-2_6
- https://reference.medscape.com/article/926678-overview#a1
- https://rarediseases.org/rare-diseases/alagille-syndrome/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5736143/
- https://medlineplus.gov/genetics/condition/alagille-syndrome/#causes